![Logo: Junge Dermatologen im Berufsverband der deutschen Dermatologen - zur Startseite [ALT+1]](/typo3conf/ext/judermconfig/Resources/Public/Images/Branding/JuDermLogo.png?v=2)
Diagnose
31. März 2022
Diagnose:
Blasenbildende kutane Porphyrie
Diskussion
Bei den Porphyrien handelt es sich um Stoffwechselstörungen, die in den meisten Fällen durch einen monogen vererbten Funktionsverlust von Enzymen der Hämbiosynthese verursacht werden. Diese Enzymdefekte führen zur Akkumulation eines oder mehrerer Intermediärmetaboliten und letztendlich zu einer signifikanten Verringerung des Endprodukts Häm [1].
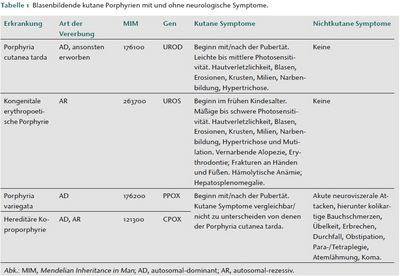
Die Porphyrien können in kutane und nichtkutane Formen eingeteilt werden. Innerhalb der kutanen Formen können blasenbildende und nichtblasenbildende Varianten unterschieden werden. Zur ersten Gruppe gehören die Porphyria cutanea tarda (PCT) [2], die Pseudoporphyria cutanea tarda [3], die Porphyria variegata (PV) [4], die hereditäre Koproporphyrie (HCP) und die kongenitale erythropoetische Porphyrie. Während sich die kongenitale erythropoetische Porphyrie in der frühen Kindheit manifestiert, entwickeln sich die anderen vier Formen nicht vor der Pubertät. Die PCT und die Pseudoporphyria cutanea tarda unterscheiden sich von der PV und der HCP durch das mögliche Auftreten akuter neuroviszeraler und psychiatrischer Symptome bei den beiden letztgenannten Varianten. Insbesondere können Patienten mit PV und HCP langanhaltende kolikartige Bauchschmerzen entwickeln, wie auch bei unserer Patientin (Tabelle 1) [1]. Diese akuten Porphyrie- Attacken werden durch Triggerfaktoren wie porphyrinogene Medikamente, Hormone oder Alkohol ausgelöst [5]. Da sich die Symptome bei unserer Patientin während ihrer ersten Schwangerschaft entwickelten und im Rahmen ihrer Menstruation rezidivierten, vermuteten wir eine hormonelle Triggerung.
Um die PV von der HCP zu differenzieren, führten wir einen fluorometrische Analyse der Plasmaporphyrine und eine biochemische Analyse der Urin- und Stuhlporphyrine durch. In der Plasmafluoreszenzanalyse zeigte sich ein Emissionsmaximum bei 625 nm.Die Stuhlanalyse wies erhöhte Mengen an Protoporphyrin und Koproporphyrin auf, wobei mehr Protoporphyrin als Koproporphyrin nachweisbar war. Diese Ergebnisse wiesen auf eine PV hin, die nachfolgend durch eine molekulargenetische Analyse bestätigt werden konnte [4, 6].
Die PV (MIM 176200) wird autosomal-dominant vererbt und wurde erstmalig im Jahr 1937 von van den Bergh und Grotepass beschrieben [7]. In Europa liegt die geschätzte Prävalenz bei etwa 1 : 100 000 [5].
Klinisch manifestiert sich die PV in der Regel mit/nach Beginn der Pubertät. Es entwickeln sich Blasen, Erosionen, Krusten, Milien und hypo-/hyperpigmentierte Narben, die auf die lichtexponierten Körperareale beschränkt sind, so wie bei unserer Patientin (Abbildung 1) [4, 6]. Wie ebenfalls bei unserer Patientin zu beobachten, können die Betroffenen eine moderate bis ausgeprägte Hypertrichose ausbilden (Abbildung 2) [6]. Darüber hinaus können akute neuroviszerale und psychiatrische Symptome auftreten, hierunter kolikartige Bauchschmerzen, Übelkeit, Erbrechen, Para-/Tetraplegie, Atemlähmung und respiratorisches Koma [1, 5, 8]. Eine mögliche Komplikation ist die Entstehung eines hepatozellulären Karzinoms [9].
Die Differenzialdiagnose umfasst andere Formen blasenbildender kutaner Porphyrien (Tabelle 1) und die akute intermittierende Porphyrie, die häufigste Form der akuten Porphyrien, die jedoch nicht mit Hautveränderungen einhergeht [1, 5, 6]. Während einer akuten Porphyrie-Attacke findet sich üblicherweise ein Anstieg der Porphyrinvorstufen δ Aminolävulinsäure und Porphobilinogen im Urin.
Zu den weiteren Charakteristika der PV gehören bei klinisch symptomatischen Betroffenen ein Plasmafluoreszenzemissionsmaximum bei 625 bis 626 nm [2] und erhöhte Mengen an Protoporphyrin und Koproporphyrin im Stuhl [1, 6] wie bei unserer Patientin. Die PV wird durch Mutationen im Protoporphyrinogenoxidase (PPOX)-Gen hervorgerufen [1, 5, 6], weshalb eine molekulargenetische Analyse die spezifischste Diagnostik darstellt.
Prophylaktisch und therapeutisch kann die Entwicklung von Hautveränderungen durch photoprotektive Kleidung, (Sonnen)Lichtschutz und die Vermeidung potenzieller Triggerfaktoren positiv beeinflusst werden. Die Entwicklung einer akuten Porphyrie-Attacke sollte dazu veranlassen, potenzielle Triggerfaktoren ausfindig zu machen und zu eliminieren. Gegebenenfalls kann initial eine intensivmedizinische Überwachung erforderlich werden. In der Behandlung sollten Häm-Präparate intravenös verabreicht werden, entweder Häm Arginat (Normosang®; nur in Europa erhältlich) oder Hämin (Panhematin®; nur in den USA erhältlich) [5]. Unter einer supportiven Therapie mit adäquater Kontrolle von Schmerzen, Übelkeit und Erbrechen klingen die Beschwerden in der Regel innerhalb von 48 bis 72 Stunden ab.
Zusammenfassend sollte bei Patienten mit auf die (sonnen) lichtexponierten Körperareale beschränkten blasigen Hautveränderungen stets eine kutane Porphyrie ausgeschlossen
werden. Es ist wichtig, die mögliche Entwicklung potenziell lebensbedrohlicher akuter Porphyrie-Attacken zu berücksichtigen. Falls erforderlich, sollten die Betroffenen interdisziplinär
behandelt werden.
Danksagung
W.M. hat ein Reisestipendium für die Teilnahme am Kongress “Dermatologie KOMPAKT & PRAXISNAH 2020” in Dresden erhalten. M.A.H. wurde u.a. durch ein Stipendium des Göttinger Promotionskollegs für Medizinstudierende unterstützt, das von der Else Kröner-Fresenius-Stiftung (2017_Promotionskolleg.04) vergeben wurde. Er wurde des Weiteren durch ein Stipendium der Deutschen Stiftung Dermatologie und ein Stipendium der Studienstiftung des deutschen Volkes gefördert. Darüber hinaus erhielt er das Kurt und Eva Herrmann-Stipendium 2019, vergeben von der Alfred-Marchionini-Stiftung.
Interessenkonflikt
Keiner.
Literatur
1 Karim Z, Lyoumi S, Nicolas G et al. Porphyrias: A 2015 update. Clin Res Hepatol Gastroenterol 2015; 39: 412–25.
2 Langer E, Weidenthaler-Barth B, Steinbrink K. Blasenbildung an den Händen nach Maniküre in einem Nagelstudio. J Dtsch Dermatol Ges 201; 16: 1058–60.
3 Weidner T, Tittelbach J, Schliemann S et al. Blasen, Ulzera, Krusten und Atrophien an Handrücken und Unterarmstreckseiten. J Dtsch Dermatol Ges 2018; 16: 88–91.
4 Collantes-Rodríguez C, de la Varga-Martínez R, Villegas- Romero I et al. Zwei neue Mutationen im PPOX-Gen bei einem Patienten mit Porphyria variegata. J Dtsch Dermatol Ges 2020; 18: 381–3.
5 Siegesmund M, van Tuyll van Serooskerken AM, Poblete-Gutierrez P et al. The acute hepatic porphyrias: current status and future challenges. Best Pract Res Clin Gastroenterol 2010; 24: 593–605.
6 Frank J, Christiano AM. Variegate porphyria: past, present and future. Skin Pharmacol Appl Skin Physiol 1998; 11: 310–20.
7 van der Bergh AAH, Grotepass W. Ein bemerkenswerter Fall von Porphyrie. Wien Klin Wochenschr 1937; 50: 830–1.
8 Mustajoki P. Variegate porphyria. Ann Intern Med 1978; 89: 238–44.
9 Schneider-Yin X, van Tuyll van Serooskerken AM, Went P, Tyblewski W et al. Hepatocellular carcinoma in variegate porphyria: a serious complication. Acta Derm Venereol 2010; 90:
512–5.
Die Diagnosequizze werden uns freundlicherweise zur Verfügung gestellt vom
"Journal der Deutschen Dermatologischen Gesellschaft" © Deutsche Dermatologische Gesellschaft