![Logo: Junge Dermatologen im Berufsverband der deutschen Dermatologen - zur Startseite [ALT+1]](/typo3conf/ext/judermconfig/Resources/Public/Images/Branding/JuDermLogo.png?v=2)
Diagnose
21. November 2024
Diagnose: Granulomatosemit Polyangiitis
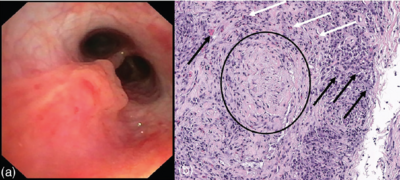
Bronchoskopie. (b) Biopsat der Bronchialwand: lymphoplasmazelluläres Entzündungsinfiltrat mit Russell-Körperchen (schwarze Pfeile), Eosinophilen (weiße Pfeile) und einem obliterierten Gefäß (schwarzer Kasten). (Hämatoxylin-Eosin-Färbung, Maßstabsbalken: 100 μm).
WEITERE BEFUNDE
Laborchemisch fand sich eine leicht erhöhte BSG bei normwertigem CRP. In der Immunserologie fielen positive PR3-ANCAs auf, die restliche Labordiagnostik war unauffällig. Die schmerzhaften Knochenveränderungen (Vorfuß, Becken, Ellbogen) wurden in Korrelation von Klinik und MRT-Bildgebung als CRMO, unabhängig von der Granulomatose mit Polyangiitis (GPA), eingeordnet. Bei einer Bildgebung im Rahmen der CRMO fiel zufällig ein pulmonaler Rundherd im rechten Oberlappen auf, der sich im MRT-Thorax als zystisch-eingeblutete Läsion darstellte. Zusätzlich zeigten sich multiple, einschmelzende Nodi beider Lungenoberlappen und prominente perihiläre Lymphknoten. Endobronchiale Biopsien zeigten ein granulomatöses Entzündungsinfiltrat mit Eosinophilen und obliterierte, intraparenchymale, kleine arterielle Gefäße (Abbildung 3). Bei rezidivierender Otitis externa rechtsseitig mit blutigeitriger Otorrhoe zeigte sich eine krustös belegte Schleimhautschwellung des äußeren Gehörgangs; im CT fand sich eine Knochenarrosion am Gehörgangsboden. Es erfolgte eine lokale Steroid- und Antibiotikatherapie und der Gehörgang wurde wiederholt fachärztlich gereinigt. Ein interdisziplinäres Entzündungsboard (Rheumatologie, Pädiatrie, Dermatologie) stellte schließlich die Diagnose einer GPA.
KLINISCHER VERLAUF
Zur Remissionsinduktion bei generalisierter Erkrankungmit Lungenbeteiligung wurde Methotrexat 15 mg/Woche s.c. kombiniert mit 45 mg Prednisolon p.o. (1 mg/kg Körpergewicht) mit einem Ausschleichschema über Monate eingeleitet. Weiterhin erfolgten vier Rituximabzyklen (540 mg i.v. an Tag 1 und 7). Zudemwurde Cotrimoxazol 960mg/Woche als Infektionsschutz (Pneumocystis jirovecii) verabreicht. Aktuell zeigt sich hinsichtlich der GPA ein stabiler Befund, die Patientin hat keine systemischen Symptome oder funktionellen Einschränkungen. Die CMRO kam im Verlauf klinisch und radiologisch zum Stillstand. Die Hautveränderungen sind narbig abgeheilt. Zur Remissionserhaltung werden aktuell MTX 7,5mg/Woche und Prednisolon 5 mg/d verabreicht.
DISKUSSION
Die GPA (früher: Wegener-Granulomatose) ist eine rheumatologische Krankheit, die mit nekrotisierender, ANCA assoziierter Vaskulitis der kleinen bis mittelgroßen Gefäße sowie granulomatöser Entzündung des Respirationstrakts einhergeht. Die Erkrankung manifestiert sich meist im 40.–50. Lebensjahr, ohne Geschlechtsprädilektion (1). Das lokalisierte Stadium beginnt typischerweise mit einer Entzündung des oberen Respirationstrakts mit oralen oder nasalen Ulzera mit eitrig-blutiger Sinusitis und Rhinitis sowie chronischer Otitis. Häufig leiden die Patienten auch an Fieber, nicht selten auch an Arthralgien und Arthritiden. Meistens kommt es im generalisierten Stadium im Verlauf zu systemischen Vaskulitis Manifestationen mit Beteiligung von Niere (nekrotisierende Glomerulonephritis) und/oder Lunge (pulmonale, nicht verkäsende Granulome und/oder alveoläre Hämorrhagien). In circa 20% der Erkrankungsfälle finden sich Hautmanifestationen, hier sind unter anderem Ulzerationen, palpable Purpura, akrale Nekrosen, akneiforme Papeln und subkutane Nodi beschrieben, die klinisch oft unspezifisch sind und an andere Entitäten denken lassen (1, 2).
Das Vorkommen von antineutrophilen zytoplasmatischen Antikörpern (ANCA) legt einen pathogenetischen Autoimmunmechanismus nahe. Antineutrophile zytoplasmatische Antikörper haben das Potenzial durch Freisetzung von reaktiven Sauerstoffradikalen und Proteasen zu einer nekrotisierenden Vaskulitis zu führen. Begleitend zur Vaskulitis bilden sich Granulome. Der Prozess der Granulombildung ist nicht vollständig geklärt; möglicherweise liegt eine T-Zell-Hyperaktivität vor (3). Bei 90% der Patienten im generalisierten Stadium kann ein positiver cANCA (PR3) nachgewiesen werden, der mit der Krankheitsaktivität korreliert (3).
Beim Vorliegen einer serologisch und histologisch gesicherten ANCA-assoziierten Vaskulitis, welche eine Subgruppe der primären systemischen Vaskulitiden nach der Chapel Hill consensus conference darstellt, kommen je nach klinisch-pathologischen Merkmalen folgende Differenzialdiagnosen in Betracht: GPA, eosinophile Granulomatose mit Polyangiitis (EGPA) und mikroskopische Polyangiitis (MPA) (4,5). Bei der differenzialdiagnostischen Abwägung im vorliegenden Falle wurde insbesondere berücksichtigt, dass bei der MPA granulomatöse Organinfiltrate fehlen und bei der EGPA eine Bluteosinophilie und ein Asthma vorliegen (4), was bei unserer Patientin nicht der Fall war. Anhand der im Jahr 2022 revidierten Klassifikationskriterien nach ACR/EULAR ergibt sich im vorliegenden Fall ein kumulativer Punktewerte von 9 (positive PR3-ANCAs, pulmonaler Rundherd in der thorakalen Bildgebung sowie eine granulomatöse Entzündung in der Histologie). Bei Diagnose einer Klein-/Mittelgefäßvaskulitis erlaubt ein Cut-off-Wert von ≥ 5 Punkten die Klassifikation als GPA (6).
Entscheidend sind Biopsien der betroffenen Organsysteme, wobei sich bei der GPA häufig Bindegewebsnekrosen umgebende Palisadengranulome und mehrkernigen Riesenzellen sowie vaskulitisch veränderte Gefäße zeigen (2, 4). Die Therapie richtet sich nach Stadium und Aktivität der Krankheit. Zur Remissionsinduktion werdenbei aktiver GPA, die nicht Organ- oder lebensbedrohlich ist, entweder Rituximab (RTX) oder Methotrexat (MTX) jeweils kombiniert mit Glukokortikoiden (GC) empfohlen, während für Patienten mit Organ- oder lebensbedrohlichem Verlauf RTX oder Cyclophosphamid (CYC) kombiniert mit GC empfohlen wird (5). Als relativ neue Empfehlung zur Remissionsinduktion kann auch der Einsatz des oralen C5aR-Inhibitors Avacopan kombiniert mit RTX oder CYC erwogen werden, um die GC-Dosen zu reduzieren (5).
Im generalisierten Stadium mit Organversagen (zumeist Nierenversagen) wird additiv zu CYC plus GC ein Plasmaaustausch empfohlen (5). Zum Remissionserhalt wird primär RTX empfohlen (Therapiedauer: 24–48 Monate); alternativ MTX oder Azathioprin (5). Die Therapie bei refraktären Verläufen bleibt herausfordernd. Studien zeigten eine gute Effektivität von RTX bei refraktären Vaskulitis Manifestationen, bei einer hohen Rate an Nonrespondern bezüglich granulomatöser Manifestationen (7).
Vom Auftreten der ersten Manifestationen bis zur Diagnosestellung und entsprechender Therapieeinleitung sind circa 4 Jahre vergangen. Die Diagnose einer GPA wird heutzutage durchschnittlich nach 4 Monaten gestellt, demnach hat die Diagnosefindung in unserem Fall außergewöhnliche lange gedauert (8).
Bei unklaren Hautveränderungen von variabler Klinik, insbesondere wenn sich histologisch eine granulomatöse Dermatitis zeigt, sollte gezielt nach extrakutanen Symptomen aus dem HNO-, nephrologischen und pulmonologischen Fachgebiet gefragt werden, um eine GPA nicht zu übersehen.
INTERESSENKONFLIKT
Keiner.
LITERATUR
- Hu CH, O’Loughlin S, Winkelmann RK. Cutaneous manifestations of Wegener granulomatosis. Arch Dermatol.1977;113:175-182.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener’s granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. Journal of Cutaneous Pathology. 2007;34:739-747.
- Tee QX, Wong A, Nambiar M, et al. Granulomatosis with polyangiitis: Common and uncommon presentations. J Med Imaging Radiat Oncol. 2022;66:1089-1096.
- Sunderkötter CH, Zelger B, Chen KR, et al. Nomenclature of Cutaneous Vasculitis. Arthritis Rheumatol. 2018;70:171-184.
- Hellmich B, Sanchez-Alamo B, Schirmer JH, et al. EULAR recommendations for themanagement of ANCA-associated vasculitis: 2022 update. Ann RheumDis.2024;83(1):30-47.
- Robson JC, Grayson PC, Ponte C, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for granulomatosis with polyangiitis. Ann Rheum Dis. 2022;81(3):309-314.
- Holle JU, Dubrau C, Herlyn K, et al. Rituximab for refractory granulomatosis with polyangiitis (Wegener’s granulomatosis): comparison of efficacy in granulomatous versus vasculiticmanifestations. Ann Rheum Dis. 2012;71(3):327-33.
- Takala JH, Kautiainen H, Malmberg H, Leirisalo-Repo M. Wegener’s granulomatosis in Finland in 1981–2000: clinical presentation and diagnostic delay. Scand J Rheumatol. 2008;37(6), 435-438.

Die Diagnosequizze werden uns freundlicherweise zur Verfügung gestellt vom
"Journal der Deutschen Dermatologischen Gesellschaft" © Deutsche Dermatologische Gesellschaft