![Logo: Junge Dermatologen im Berufsverband der deutschen Dermatologen - zur Startseite [ALT+1]](/typo3conf/ext/judermconfig/Resources/Public/Images/Branding/JuDermLogo.png?v=2)
Disseminierte erythematöse Papeln bei einem Patienten mit Dermatomyositis
28. Februar 2025
Maria Kinberger (1), Amrei Dilling (2), Farzan Solimani (2,3), Katharina Meier (2), Margitta Worm (4)
- Klinik für Dermatologie, Venerologie und Allergologie, Division of Evidence Based Medicine (dEBM), Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin
- Klinik für Dermatologie, Venerologie und Allergologie, Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin
- BIH Biomedical Innovation Academy, BIH Charit. Clinician Scientist Program, Berlin Institute of Health at Charité – Universit.tsmedizin Berlin
- Klinik für Dermatologie, Venerologie und Allergologie, Abteilung für Allergologie und Immunologie, Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin

perivaskulär betontes, vorwiegend lymphozyt.res Infiltrat (H.matoxylin-Eosin-Färbung, Maßstabsbalken: 200 μm).
ANAMNESE UND KLINISCHER BEFUND
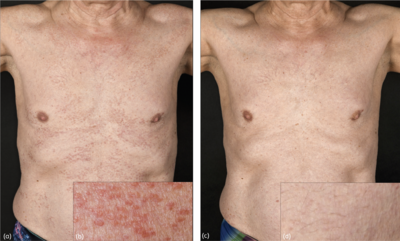
Wir berichten über einen 63-jährigen, kaukasischen Patienten, der sich auf Anraten seines behandelnden Rheumatologen in unserer dermatologischen Klinik vorstellte. In rheumatologischer Behandlung befand sich der Patient aufgrund einer anti-MDA5-Dermatomyositis, die nach dem Auftreten einer proximalen Muskelschwäche und Myalgien in den Schultern, Oberschenkeln und im Gesäßbereich vor einigen Monaten diagnostiziert und mittels Muskelbiopsie und serologischen Untersuchung bestätigt worden war. Diesbezüglich erhielt der Patient eine Therapie mit systemischen Prednison (initial 25 mg pro Tag, mit anschließender Reduzierung bis auf 2 mg). Als suspekt wertete der behandelnde Arzt jedoch disseminierte, stammbetonte erythematöse Papeln, die nicht dem klassischen Hautbefund einer Dermatomyositis entsprachen. Die Anamnese ergab, dass der Patient bereits seit 5 Jahren unter diesen papulösen Läsionen litt (während die Symptome der Dermatomyositis erst einige Monate zuvor aufgetreten waren). Da die Läsionen jedoch weder Juckreiz noch andere Beschwerden verursacht hatten, war bislang keine Abklärung erfolgt. Bei der klinischen Untersuchung zeigten sich im Bereich des Stammes und der Extremitäten disseminierte erythematöse Papeln ohne epidermale Beteiligung (Abbildung 1a,b). Sonstige Dermatomyositis-typische Hautbefunde wie heliotrope Erytheme, Lipödeme, Gottron-Papeln, oder für die MDA5-Antikörper positive Dermatomyositis typische Hautbefunde wie Ulzerationen, Calcinosis oder Alopezie zeigten sich nicht.
HISTOPATHOLOGIE
In der histologischen Untersuchung zeigten sich in der oberen Dermis umschriebene Granulome aus epitheloidzelligen Makrophagen und mehrkernigen Riesenzellen mit abschnittsweise zentraler Nekrose. Im Randbereich der Granulome zeigten sich einige Lymphozyten sowie oberflächlich perivaskulär ein lymphohistiozytäres Infiltrat mit beigemengten Plasmazellen. Vermehrtes Muzin ließ sich in der oberen Dermis interstitiell nachweisen (Abbildung 2). Die Granulome wiesen dementsprechend sowohl sarkoidale als auch tuberkuloide Merkmale auf.
WEITERE DIAGNOSTIK
Das Differenzialblutbild zeigte sich bis auf eine milde Lymphopenie (1,02/nl) unauffällig. Auch der Kreatinkinase-Wert (CK) zeigte sich normwertig. In der klinischen Chemie ergaben sich weitere Normwerte für den löslichen Interleukin-2 Rezeptor und für das angiotensin converting enzyme (ACE). Eine Lues-Serologie sowie ein Quantiferontest waren negativ. Eine PCR-Untersuchung auf Mykobakterien aus der Probebiopsie fiel ebenfalls negativ aus.
KLINISCHER VERLAUF
Aufgrund der Dermatomyositis erfolgte im Verlauf die Einleitung einer Therapie mit Methotrexat p.o. (initial 10 mg pro Woche, welche bei guter Verträglichkeit im Verlauf auf 15 mg pro Woche gesteigert werden konnte) (1). Neben dem Abklingen der Myositis-Symptome kam es auch zu einer nahezu vollständigen Remission des Hautbefundes binnen 8 Monaten (Abbildung 1c,d).
Ihre Diagnose?…