![Logo: Junge Dermatologen im Berufsverband der deutschen Dermatologen - zur Startseite [ALT+1]](/typo3conf/ext/judermconfig/Resources/Public/Images/Branding/JuDermLogo.png?v=2)
Diagnose
23. September 2024
Diagnose: Palmoplantarkeratose – kongenitale Alopezie (PPKCA)-Syndrom Typ 2
KLINISCHER VERLAUF
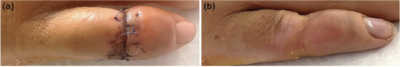
Der Schnürring wurde mikrochirurgisch reseziert und der Finger mit primärem Wundverschluss rekonstruiert (Abbildung 2a). Ein intraoperativer Abstrich ergab Staphylococcus aureus. Die Patientin erhielt für 5 Tage Ampicillin/Sulbactam intravenös, gefolgt von Amoxicillin/Clavulansäure per os für 10 Tage. Das Nahtmaterial wurde nach 15 Tagen entfernt, und die Wunde heilte reizlos ab (Abbildung 2b). Das palmoplantare Keratoderm (PPK) wurde mit 10% Urea und mit mechanischer Keratolyse behandelt. Orale Retinoide, zum Beispiel Alitretinoin, wurden angeboten, derzeit jedoch nicht gewünscht. Lebenslange dermatologische und handchirurgische Folgeuntersuchungen und eine rechtzeitige Resektion weiterer Schnürringe wurden empfohlen.
GENETISCHE ANALYSE
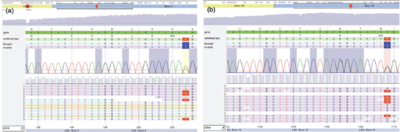
Eine Exom-Analyse mit Next Generation Sequencing erbrachte zwei heterozygote Varianten im Lanosterol-Synthase (LSS)-Gen, eine Variante unklarer Signifikanz (Klasse 3) in Exon 3 (c.207A>T; p.(Lys69Asn)) (Abbildung 3a) und eine wahrscheinlich pathogene Variante (Klasse 4) in Exon 19 (c.1769G>A; p.(Trp590*)) (Abbildung 3b; Referenz-Sequenz NM_001001438.2, GRCh37.p13). Blut oder DNA des Vaters der Patientin war nicht für genetische Untersuchungen verfügbar. Eine Blutprobe der Mutter wird wahrscheinlich zu einem späteren Zeitpunkt für eine Segregationsanalyse zur Verfügung gestellt.
DISKUSSION
Das PPK-CA-Syndrom Typ 2 ist ein sehr seltenes, autosomal rezessiv vererbtes palmoplantares Keratoderm, das durch lineare transgrediente Hyperkeratosen an den medialen und lateralen Kanten der Hände, Finger, Füße und Zehen, progressive Einschnürungen, Sklerodaktylie, milde Nageldystrophie, Ulerythema ophryogenes, Keratosis pilaris und eine kongenitale Alopecia universalis charakterisiert ist. Die ersten Fälle wurden 1989 von Wallis et al. (1) und Bhatia et al. (2) beschrieben. Das Syndrom wurde 2010 von Castori et al. als Entität klassifiziert und vom milderen, autosomal dominant vererbten PPK-CA Typ 1 unterschieden (3). Von dieser Gruppe wurde auch der Phänotyp unserer Patientin im Alter von 10 und 16 Jahren beschrieben (3,4). Damals konnte jedoch trotz umfangreicher Analysen keine verursachende Genvariante nachgewiesen werden (3,4). Yang et al. identifizierten kürzlich biallelische Varianten im LSS-Gen als wahrscheinliche Ursache für PPK-CA Typ 2 bei zwei nicht verwandten Han-chinesischen Patienten. Beide Patienten litten zusätzlich unter einem Katarakt, einer wies eine Agenesie des Corpus callosum und eine milde intellektuelle Beeinträchtigung auf (5). Ex-vivo-Studien ergaben eine Herunterregulierung und eine vermindere enzymatische Aktivität der LSS-Varianten. Immunfluoreszenzund Transmissionselektronenmikroskopie zeigten einen gestörten Aufbau des cornified envelope mit unregelmäßig geformten Lipidlamellen (5). Außerdem beschrieben Wang und Kollegen zwei Familien mit PPK und Hypotrichose mit LSS-Genvarianten (6). Zhou et al. berichteten über einen chinesischen Patienten mit biallelischen LSS-Mutationen und mutilierendem PPK ohne Alopezie, der erfolgreich mit 2% Cholesterol 2% Simvastatin-Creme behandelt wurde (7). Die Lanosterol-Synthase ist an der Cholesterinbiosynthese beteiligt, bei der es (S)-2,3-Oxidosqualen in Lanosterol konvertiert. Es wurde spekuliert, dass ein Ungleichgewicht dieser Lipide aufgrund von LSS-Genvarianten zu einer gestörten Anlagerung und Quervernetzung der lamellar bodies (Odland-Körperchen) und zu einer abnormen Anordnung der Lipide im cornified envelope führt (5). Die bei unserer Patientin nachgewiesene LSS-Variante c.207A>T bewirkt einen Austausch von Lysin durch Asparagin an Aminosäure-Position 69. Die Variante c.1769G>A führt zu einem prämaturen Stopcodon an Position 590. Diese trunkierte Variante kann einen nonsense-mediated mRNA Decay zur Folge haben, der die Bildung eines trunktierten LSS-Proteins verhindert. Beide Varianten waren in öffentlichen Datenbanken nicht eingetragen. LSS-Variantenwurden auch bei autosomal rezessiver kongenitaler Alopezie mit intellektueller Beeinträchtigung (8), autosomal rezessiver Hypotrichosis simplex (9) und kongenitalen Katarakten beschrieben (10). Demnach haben Varianten in diesem Gen eine große phänotypische Variabilität. Genotyp-Phänotyp-Korrelationen müssen etabliert werden, sobald eine ausreichende Anzahl an Fällen verfügbar ist.
DANKSAGUNG
Wir danken der Patientin für die Zustimmung zur Publikation ihres Falls.
INTERESSENKONFLIKT
Keiner
LITERATUR
- Lubeck G, Epstein E. Complications of tattooing. Calif Med.1952;76(2):83-85.
- Kluger N. Tattoo-associated uveitis with or without systemic sarcoidosis: a comparative review of the literature. J Eur Acad Dermatol Venereol. 2018;32(11):1852-1861.
- Mansour, AM, Chan CC. Recurrent uveitis preceded by swelling of skin tattoos. AmJ Ophthalmol. 1991;111(4):515-516.
- RorsmanH, Brehmer-Andersson E, Dahlquist I, et al. Tattoo granuloma and uveitis. Lancet. 1969;2(7610):27-28.
- Larese Filon F,Mauro M, AdamiG, et al. Nanoparticles skin absorption: New aspects for a safety profile evaluation. Regul Toxicol Pharmacol. 2015;72(2):310-22.
- Weiss KT, Schreiver I, Siewert K, et al. Tattoos – more than just colored skin? Searching for tattoo allergens. J Dtsch Dermatol Ges. 2021;19(5):657-669.
- Sepehri M, Hutton Carlsen K, Serup J. Papulo-Nodular Reactions in Black Tattoos as Markers of Sarcoidosis: Study of 92 Tattoo Reactions from a Hospital Material. Dermatology. 2016;232(6):679-686. 16100387, 2024, 7, Downloaded from onlinelibrary.wiley.com/doi/10.1111/ddg.15414_g by Ute Pellmann , Wiley Online Library on [17/07/2024]. See the Terms and Conditions (https://onlinelibrary.wiley.com/terms-and-conditions) on Wiley Online Library for rules of use; OA articles are governed by the applicable Creative Commons License
- Ungprasert P, Ryu JH, Matteson EL. Clinical Manifestations, Diagnosis, and Treatment of Sarcoidosis. Mayo Clin Proc InnovQualOutcomes. 2019;3(3):358-375.
- Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357(21):2153-2165.
- Kwaczala-Tessmann A, Dellasette F, Brix A, et al. Fremdkörpergranulome im Bereich einer Tätowierung: Tattoo-assoziierte Uveitis als Wegweiser zur Diagnose einer Sarkoidose? J Dtsch Dermatol Ges. 2023;21(Suppl. 2):31-33.
Die Diagnosequizze werden uns freundlicherweise zur Verfügung gestellt vom
"Journal der Deutschen Dermatologischen Gesellschaft" © Deutsche Dermatologische Gesellschaft